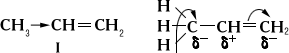|
|
Популярные авторы:: БСЭ :: Борхес Хорхе Луис :: Горький Максим :: Азимов Айзек :: Херберт Фрэнк :: Чехов Антон Павлович :: Раззаков Федор :: Толстой Лев Николаевич :: Грин Александр :: Астафьев Виктор Петрович Популярные книги:: Дюна (Книги 1-3) :: The Boarding House :: Три цвета времени :: Упражнения по стилистике русского языка :: Преодоление :: Талисман из Рэдволла :: Плацдарм :: Закрой рот, джинн – кишки простудишь… :: Авантюристка :: Андропов. 7 тайн генсека с Лубянки |
Большая Советская Энциклопедия (ОР)ModernLib.Net / Энциклопедии / БСЭ / Большая Советская Энциклопедия (ОР) - Чтение (стр. 7)
Начиная с середины 19 в. число органических веществ, получаемых синтетически, быстро возрастает. Так, в 1842 Н. Н.
восстановлением нитробензола получил анилин, в 1845 А.
синтезировал уксусную кислоту, в 1854 П.
- вещества типа жиров. В 1861 А. М.
получил первое искусственное сахаристое вещество, названное им метиленитаном, из которого впоследствии была выделена акроза. Синтетическое направление в О. х. приобретает всё большее значение. В результате успехов синтеза господствовавшее идеалистическое представление о необходимости «жизненной силы» для создания органических веществ (см.
) было отвергнуто.
Теоретические представления в О. х. начали развиваться со 2-й четверти 19 в., когда была создана (Либих, Вёлер, Э. , Р. и др.). Основное её положение о переходе группы атомов - радикалов из одного соединения в другое в неизменном виде остаётся в большом числе случаев справедливым и в настоящее время. На этом представлении основаны многие физические и химические методы исследования веществ неизвестной структуры. Впоследствии (1834-39) Дюма показал возможность замещения положительно заряженных атомов в радикале на электроотрицательные без серьёзных изменений электрохимического характера радикала, что до Дюма считалось невозможным. На смену теории радикалов пришла (1848-51, 1853), созданная Дюма, Ш. и О. . Последним удалось классифицировать органические вещества по типам простейших неорганических соединений. Так, спирты считались соединениями типа воды, амины - типа аммиака, галогеналкилы - типа хлористого водорода. Позднее Ф. А. установил четвёртый тип - тип метана, от которого он производил все углеводороды. Теория типов позволила создать чёткую классификацию органических соединений, которая лежит в основе современной классификации органических веществ. Однако эта теория стремилась лишь к объяснению реакционной способности органических веществ и отрицала принципиальную возможность познания их строения. В 1853 Франкленд, изучая металлоорганические соединения, ввёл представление о валентности. В 1857 Кекуле высказывает мысль о возможности сцепления атомов углерода друг с другом и доказывает четырёхвалентность углерода. В 1858 А. , используя правило валентности и положение Кекуле о сцеплении атомов углерода, впервые отходит от теории типов и пишет формулы органических веществ, очень близкие к современным. Однако идеи теории типов оставались ещё очень сильны и создание теории продолжало отставать от развития эксперимента. В 1861 Бутлеров создал органических веществ. Он ввёл в О. х. ряд новых понятий: о химической связи, порядке связей атомов в молекуле, о взаимном влиянии атомов, непосредственно связанных или не связанных друг с другом, и др. Теория строения Бутлерова блестяще объяснила остававшиеся непонятными известные к тому времени случаи изомерии. В 1864 Бутлеров предсказал возможность изомерии углеводородов и вскоре (1867) подтвердил это синтезом изобутана. Созданное Бутлеровым стройное учение лежит в основе современных представлений о химической строении органических веществ. Одно из важнейших положений теории строения - о взаимном влиянии атомов - впоследствии было развито В. В. . Детальное изучение этого влияния способствовало дальнейшему развитию теории строения и представлений о распределении электронной плотности и о реакционной способности органических соединений. В 1869 И. показал, что явление изомерии наблюдается и при совершенно одинаковой последовательности сцепления атомов в молекуле. Он доказал идентичность строения обычной и мясо-молочной и пришёл к выводу, что тонкие различия в свойствах молекул с одинаковой структурой следует искать в различном расположении их атомов в пространстве. В 1874 Я. и французский химик Ж. Ле Бель создали теорию пространств. расположения атомов в молекуле - . В основе этой теории, по Вант-Гоффу, лежит представление о тетраэдрической модели четырёхвалентного атома углерода и о том, что оптическая изомерия является следствием пространственной асимметрии молекулы, в которой атом углерода соединён с четырьмя различными заместителями (см. ). Вант-Гофф высказал также предположение о возможности др. вида пространственной изомерии при отсутствии в молекуле асимметричного атома углерода. Вскоре Вислиценус доказал, что фумаровая кислота, которую ранее считали полимером малеиновой кислоты, представляет собой её геометрический изомер (геометрическая, или -изомерия). Ясно, что стереохимическое учение могло быть создано только на основе представлений о строении (структуре) молекулы в бутлеровском понимании. К конце 19 в. накопился большой фактический материал, в том числе и по ароматическим соединениям; в частности, широко была изучена химия бензола, открытого М. в 1825. Первая т. н. «бензольная теория» строения ароматических соединений была создана в 1865 Кекуле. В ней высказывается мысль о том, что атомы углерода в органических соединениях могут образовывать кольца. Согласно этой теории, бензол обладает симметричной структурой вследствие кольцеобразного строения сцепленных попеременно простыми и двойными связями шести метиновых СН-групп. Однако, исходя из строения бензола по Кекуле, следовало допустить наличие двух орто-замещённых гомологов или производных бензола, чего на самом деле не наблюдалось. Устойчивость бензола к сильным окислителям и некоторые др. т. н. ароматическим свойства бензола и его производных также противоречили предложенной формуле. Поэтому Кекуле ввёл (1872) представление об осцилляции (быстром перемещении) двойных связей и устранил формальные различия между двумя орто-положениями. Несмотря на то, что строение бензола по Кекуле находилось в противоречии с данными о его физических и химических свойствах, оно долгое время без всяких изменений принималось подавляющим числом химиков. Т. о., остался ряд вопросов, не разрешимых с точки зрения «классической» теории строения. К этим вопросам относится и своеобразие свойств многих др. соединений с сопряжёнными системами связей. Строение бензола и др. ароматических систем могло быть установлено лишь с появлением физических методов исследования и с развитием квантово-химических представлений о строении органических веществ. Электронные представления [В. (1916) и Г. (1916)] придали физическое содержание понятию (пара обобщённых электронов); однако в том виде, в каком они были сформулированы, эти представления не смогли отразить тонких переходов от ковалентной к ионной связи и в О. х. оставались в значительной степени формальными. Только с помощью квантово-химического учения было вложено принципиально новое содержание в правильные в основном представления электронной теории. Представления Льюиса о паре электронов, образующих связь и всегда строго локализованных на этой связи, оказались приближёнными и в большинстве случаев не могли быть приняты. Современные представления теории строения и значение О. х.Учёт квантовых свойств электрона, представления об электронной плотности и о взаимодействии электронов в сопряжённых системах открыли новые возможности для рассмотрения вопросов о строении, взаимном влиянии атомов в молекуле и о реакционной способности органических соединений (см. , ). В насыщенных углеводородах одинарные связи С-С (s-связи) действительно реализуются парой электронов; в симметричных углеводородах электронная плотность в пространстве между соединившимися атомами С-С больше суммы соответствующих электронных плотностей тех же изолированных атомов и симметрично распределена относительно оси, соединяющей центры атомов. Увеличение электронной плотности - результат перекрывания электронных облаков атомов по прямой, соединяющей их центры. В несимметричных парафинах появляется возможность неполной симметрии в распределении электронной плотности; однако эта асимметрия столь незначительна, что дипольные моменты всех парафиновых углеводородов почти не обнаруживаются. То же касается и симметрично построенных непредельных углеводородов (например, этилена, бутадиена), у которых атомы С соединены друг с другом двойной связью (s- и p-связью). Введение в молекулы этих веществ электронодонорной метильной группы вследствие высокой поляризуемости p-связи приводит к смещению электронной плотности к крайнему атому углерода, и пропилен (I) уже имеет дипольный момент 0,35 Д, а 1-метилбутадиен - 0,68 Д. Распределение электронной плотности в этих случаях принято изображать одной из следующих схем:
(Знаки d +и d -показывают возникающие частичные заряды на атомах С) В представления о распределении электронной плотности хорошо укладывается ряд эмпирических правил О. х. Так, из приведённой выше формулы пропилена следует, что при гетеролитическом присоединении к нему галогеноводородов протон должен фиксироваться в месте наибольшей электронной плотности, т. е. у наиболее «гидрогенизированного» атома углерода (см. ). Значительно сильнее сказывается введение в молекулы углеводородов атомов или групп, сильно отличающихся по электроотрицательности от атомов углерода или водорода. Например, введение электрофильного заместителя в молекулы углеводородов ведёт к изменению подвижности атомов водорода в связях С-Н, О-Н и др. Подобного рода взаимное влияние атомов, также объяснимое изменением распределения электронной плотности, быстро затухает у насыщенных соединений и почти без затухания передаётся по цепи сопряжённых связей (см. ). Принято различать два вида электронного влияния заместителей: индуктивное - по цепи s-связей и влияние сопряжения - по цепи с сопряжёнными связями. Так, увеличение кислотности хлоруксусной кислоты (II) по сравнению с уксусной СН 3СООН объясняется индуктивным влиянием атомов хлора, а подвижность атомов водорода метильных групп в уксусном (III) или сорбиновом (IV) альдегиде - влиянием сопряжения:
Перераспределение электронной плотности, особенно в момент реакции, происходит не только в связях, которые затрагиваются реакцией, но и в др. частях молекулы. Кажущаяся ненормальность солеобразования n-диметиламиноазобензола (V) с фиксацией протона слабоосновным атомом азота азогруппы объясняется перемещением реакционного центра молекулы вследствие сдвига электронной плотности в момент реакции в направлении, указанном стрелками:
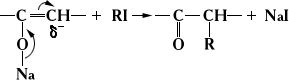
Влияние сопряжения проявляется и в тех случаях, когда два возможных направления реакции органических веществ не обусловлены . Так, алкилирование натрийенолятов по атому углерода происходит вследствие перемещения реакционного центра благодаря сопряжению связей:
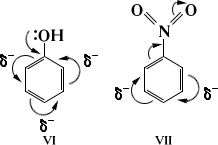
Взаимное влияние атомов в результате сопряжения связей проявляется также в ароматических соединениях (см. ). При электрофильном замещении электронодонорные (нуклеофильные) заместители (VI) ориентируют в орто- и -положения, электроноакцепторные (электрофильные) заместители (VII) - в -положение:
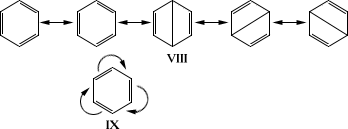
Т. о., на основании современных квантово-химических представлений разнообразные процессы О. х. нашли естественное объяснение. Теоретические представления О. х. окрепли и получили предсказательные возможности. В результате развития теоретических и физических методов исследования был окончательно решен вопрос о строении ароматических систем, в том числе и бензола. Строение последнего описывается следующим образом: шесть атомов углерода бензольного кольца находятся в одной плоскости и соединены s-связями; шесть p-электронов составляют единую подвижную электронную систему. Следствием этого является полная подтверждаемая опытом равноценность связей С-С и высокая симметрия бензола с осью шестого порядка. Из этих положений следует, что бензол неполярен и обладает анизотропией диамагнитной восприимчивости. Аналогичными свойствами характеризуются все ароматические системы, у которых число p-электронов равно 4 n+ 2 (правило Хюккеля). Бензол - далеко не единичный пример соединений с выравненными двойными и простыми связями; аналогичная картина наблюдается у трополона, тропилийбромида, ферроцена, дифенилполиенов и др. Вполне удачного графического изображения строения бензола и др. ароматических соединений выработать не удалось. Для описания их строения используют набор валентных схем (VIII), впервые предложенных Л. в его , или систему обозначения (IX), где изогнутые стрелки показывают также выравненность связей (впервые применена в теории ):
Этими же обозначениями пользуются для графической интерпретации равномерного распределения электронной плотности в симметричных ионах, например в карбоксилат-анионе (соответственно Х и XI), при объяснении слабоосновных свойств амидов кислот (XII и XIII) и в др. случаях:
Приблизительно со 2-й половины 20 в. О. х. вступила в новую фазу. Многие направления её развивались столь интенсивно, что выросли в большие специализированные разделы, называется по научному или прикладному признаку (стереохимия, химия , природных веществ, , , , , , и др.). Успехи теории и развитие физических методов исследования (например, , ультрафиолетовой и , раманспектроскопии, , химически индуцированной динамической поляризации ядер, масс-спектрометрии), а также методов идентификации и разделения различных веществ с помощью сделали возможным быстрый структурный анализ сложнейших органических соединений и быстрое решение многих важных проблем. Применение физических методов для исследования кинетики реакций органических веществ (см. ) позволяет изучать реакции с периодом полупревращения 10 -8-10 -9 сек. Корреляционные уравнения, основанные на принципе линейности свободной энергии, дают возможность количественной оценки зависимостей между строением и реакционной способностью органических соединений, даже тех, которые обладают физиологическим действием. О. х. оказалась тесно связанной со смежными естественными науками - , и , применение идей и методов О. х. в этих науках в значительной степени обусловило развитие нового направления - . Методы О. х. наряду с физическими методами исследования сыграли важную роль в установлении строения нуклеиновых кислот, многих , сложных природных соединений; с их же помощью были раскрыты механизм и регуляция синтеза белков (см. ). Чрезвычайно возросли синтетические возможности О. х., которые привели к получению таких сложно построенных природных веществ, как хлорофилл, витамин B 12(Р. ), полинуклеотиды с определённым чередованием звеньев (А. , Х. Г. ) и др. Огромный успех этих методов - разработка автоматического синтеза многих полипептидов, в том числе и ферментов. Синтезирован новый класс органических соединений, образованных сплетением двух или более циклических молекул подобно обычной цепи (катенаны, на схеме слева) или подобно гантели, на ось которой надето кольцо (ротаксаны, справа):
Отдельные части этих молекул связаны механическими силами. Наиболее значительным достижением синтетической О. х. и биохимии можно считать синтез гена, который осуществил Х. Г. Корана с сотрудниками (1967-70). Большое значение приобрели методы О. х. в современной технологии производства , , , красителей, медикаментов, в промышленности кинофотоматериалов, стимуляторов роста растений, средств борьбы с вредителями сельского хозяйства ( ) и многое др. Успехи О. х. в области и не только изменили технологию ряда производств, но и привели к созданию новых видов продукции. Благодаря установлению зависимостей между строением и свойствами органических соединений становится возможным создание новых материалов различных назначений с заранее заданными свойствами. О. х. достигла такого уровня, который отвечает её важной роли в создании материальной культуры современного общества. Научные учреждения и организации, периодические издания.Научную работу по О. х. в СССР проводят научно-исследовательские институты АН СССР - институт органической химии им. Н. Д. Зелинского (ИОХ), институт органической и физической химии им. А. Е. Арбузова (ИОФХ), институт нефтехимической синтеза им. А. В. Топчиева (ИНХС), институт элементоорганич. соединений (ИНЭОС), институт химии природных соединений им. М. М. Шемякина (ИХПС); Сибирского отделения АН СССР - Новосибирский институт органической химии (НИОХ), Иркутский институт органический химии (ИИОХ), институт химии нефти; республиканскиъ академий - институты органической химии Армянской ССР, Киргизской ССР, УССР, институт тонкой органической химии им. А. Л. Мнджояна (Армянская ССР), институт физико-органической химии (БССР), институт физической и органической химии им. П. Г. Меликишвили (Грузинская ССР), институт органического синтеза (Латвийская ССР) и др. Национальный комитет советских химиков является членом Международного союза чистой и прикладной химии (International Union of Pure and Applied Chemie, IUPAC, ЮПАК), который организует 1 раз в 2 года конгрессы, конференции и симпозиумы, в частности и по О. х. Периодические издания, в которых публикуются работы по О. х.: в СССР - «Журнал органической химии» (с 1965); «Журнал общей химии» (с 1931); «Химия гетероциклических соединений» (Рига, с 1965); «Химия природных соединений» (Ташкент, с 1965); «Экспресс-информация. Промышленный органический синтез» (с 1960); за рубежом - «Journal of Organic Chemistry» (Wash., с 1936); «Journal of the Chemical Society. Perkin Transaction. 1. Organic and Bio-organic Chemistry» (L., с 1972); II. «Physical Organic Chemistry» (L., с 1972); «Justus Liebigs Annalen der Chemie» (Weinheim, с 1832); «Bulletin de la Societe 'chimique de France», pt. 2 (P., с 1858); «Journal of the Society of Organic Synthetic Chemistry of Japan» (Tokyo, с 1943); международные - «Tetrahedron» (N. Y., с 1957); «Tetrahedron Letters» (L., с 1959); «Synthesis» (Stuttgart, с 1969); «Synthetic Communication» (N. Y., с 1971); «Journal of the Organo-metallic Chemistry» (Lausanne, с 1964); «Journal of Heterocyclic Chemistry» (L., с 1964); «Organic Magnetic Resonance» (L., 1969); «Organic Mass Spectrometry» (L., 1968); «Organic Preparations and Procedures» (N. Y., с 1969). литература по О. х. реферируется в журналах: «Chemical Abstracts» (Easton, с 1907), реферативный журнал «Химия» (с 1953), «Chemisches Zentralblatt» (совместно ГДР и ФРГ, В., с 1830). Лит.:Бутлеров А. М., Введение к полному изучению органической химии, в. 1-3, Каз.,1864-66; его же, Избр. работы по органической химии, М., 1951; Марковников В. В., Избр. труды, М., 1955; Гьельт Э., История органической химии с древнейших времён до настоящего времени, пер. с нем., Хар. - К., 1937; Шорлеммер К., Возникновение и развитие органической химии, пер. с англ., М., 1937; Джуа М., История химии, пер. с итал., М., 1966; Rodd’s chemistry of carbon compounds, 2 ed., v. 1-2, Amst. - [a. o.], 1964-1968; Beilsteins Handbuch der organischen Chemie, 4 Aufl., bearb. von B. Prager [u. a.], Bd 1-34, В., 1918-1944 (с 1928 года изд. доп. тт.); Houben-Weyl, Methoden der organischen Chemie, 4 Aufl., Bd 1-12, Stuttg., 1953-68; Краткая химическая энциклопедия, т. 1-5, М., 1961-67; Несмеянов А. Н., Несмеянов Н. А., Начала органической химии, т. 1-2, М., 1969-70; Неницеску К. Д., Органическая химия, пер. с рум., т. 1-2, М., 1962-63; Робертс Дж., Касерио М., Основы органической химии, пер. с англ., ч. 1-2, М., 1968; Физер Л., Физер М., Органическая химия, пер. с англ., М., 1966; Чичибабин А. Е.., Основные начала органической химии, т. 1-2, М., 1957-63; Ингольд К., Теоретические основы органической химии, пер. с англ., М., 1973; Перспективы развития органической химии, под ред. А. Тодда, пер. с англ., М., 1959. И. Л. Кнунянц. Органическая школа Органи'ческая шко'лав социологии, направление в буржуазной социологии конца 19 - начала 20 вв., отождествлявшее общество с организмом и пытавшееся объяснить социальную жизнь биологическими закономерностями. Сравнение общества с организмом проводилось многими авторами (Платон, Т. Гоббс, О. Конт, Г. Спенсер). В отличие от своих предшественников, представители О. ш. [П. Ф. Лилиенфельд (Россия), А. Шеффле (Германия), Р. Вормс, А. Эспинас (Франция)] утверждали, что «общество и есть организм». Они подыскивали всё новые аналогии, доказывая (в различных вариантах) тождество общества и организма. Лилиенфельд приписывал обществу все черты организма - единство, целесообразность, специализацию органов. Роль кровообращения, например, выполняет торговля, функции головного мозга - правительство. Шеффле рассматривал экономическую жизнь общества как обмен веществ в организме. Вормс доходил до крайностей, рассуждая о половых различиях общественных организмов, об их органах выделения и т.д. В начале 20 в. концепции О. ш. утратили популярность. По сравнению с попытками рассматривать общество как продукт произвольного соглашения между индивидами органический подход был известным шагом вперёд. Однако в целом концепции О. ш. ненаучны. Их порок в том, что конкретно-историческое изучение социальных явлений заменялось произвольными аналогиями. Вводимые понятия были расплывчатыми, социальные закономерности подменялись биологическими, целый ряд явлений общественной жизни (конфликты, классовая борьба и т.д.) совершенно игнорировался или объявлялся «болезнями организма». Общественное развитие должно, по мнению сторонников О. ш., происходить путём эволюции. Ссылка на организмическую природу общества часто служила апологии капитализма. Марксисты употребляют термин «социальный организм». Однако в марксизме эти аналогии не подменяют конкретного изучения специфики и объективных законов общественной жизни. Лит.:Ленин В. И., Что такое «друзья народа» и как они воюют против социал-демократов?, Полн. собр. соч., 5 изд., т. 1; его же, Экономическое содержание народничества и критика его в книге г. Струве, там же; Кон И. С., Позитивизм в социологии, Л., 1964. В. Б. Ольшанский. Органические вещества почвы Органи'ческие вещества' по'чвы,комплекс органических соединений, входящих в состав . Их присутствие - один из основных признаков, отличающих почву от материнской породы. Формируются в процессе разложения растительного и животного материалов и представляют собой важнейшее звено обмена веществ живой и неживой природы. Количество О. в. п. и их природа во многом определяют направление процесса почвообразования, биологические, физические, химические свойства почвы и её плодородие. В О. в. п. входят в том или ином количестве растительные и животные остатки в различной степени разложения при обязательном преобладании гумусовых веществ (см. ). Органические вяжущие материалы Органи'ческие вя'жущие материа'лы,см. в ст. . Органические полупроводники Органи'ческие полупроводники',см. . Органические удобрения Органи'ческие удобре'ния, , содержащие питательные вещества в форме органических соединений растительного или животного происхождения. О. у. оказывают многостороннее агрономическое действие на свойства почвы. При разложении их в результате жизнедеятельности почвенных микроорганизмов образуются доступные растениям минеральные соединения N, Р, К, Са, S и др. элементов и перегной, или . Выделяющийся при этом углекислый газ насыщает почвенный воздух и приземной слой атмосферы, улучшая углеродное питание растений. При систематическом внесении О. у. улучшаются физико-химические и химические свойства почвы, её водный и воздушный режимы, активизируется жизнедеятельность полезных микроорганизмов (азот-фиксирующих бактерий, аммонификаторов и др.). Через О. у. в основном осуществляется круговорот питательных веществ по схеме: почва - растения - животные - почва. Применение О. у. позволяет вносить минеральные удобрения в больших дозах и получать высокие урожаи с.-х. культур. К О. у. относятся большинство местных удобрений ( , навозная жижа, , , птичий помёт), зелёное удобрение (см. ), отходы городского коммунального хозяйства (мусор и компосты из него, осадки сточных вод, фекальные массы), пищевой, кожевенной и др. отраслей промышленности, а также (ил), солома, и др. Содержание питательных веществ в наиболее распространённых О. у. см. в таблице. Содержание питательных веществ в органических удобрениях (в % на сухое вещество; навоз, навозная жижа, компост в % на сырое вещество)
О. у. известны с раннего периода истории земледелия. В Китае, Корее, Японии их начали применять 3 тыс. лет назад. В странах Западной Европы и на территории Европейской части СССР уже в 13-14 вв. использовали навоз, в Средней Азии с давних времён - зелёное удобрение. Ежегодное мировое применение О. у. во 2-й половине 20 в. определяется в 3-4 млрд. т, что соответствует 15-20 млн. тN, 3-4 млн. тP 2O 5и 18-24 млн. тK 2O. В СССР на поля страны было вывезено 360 млн. тО. у. в 1965, 468 млн. тв 1970, более 500 млн. тв 1973. О. у. вносят под вспашку (иногда под культивацию), в лунки при посадке (например, картофеля, капусты), в подкормку, используют как , для приготовления почвосмесей, смесей с минеральными удобрениями, для мульчирования посевов. Дозы О. у. от 15 до 60 м/га(под овощные и коноплю до 80-100 т/га), при внесении в лунки - 5-10 т/га, при применении совместно с минеральными - значительно ниже. О. у. наиболее эффективны на дерново-подзолистых почвах. Прибавки урожая ( цс 1 га): картофеля 50-60, сахарной свёклы 30, овощей 60-100, зерновых 6-7. Лит.:Справочник по удобрениям, М., 1964; Прянишников Д. Н., Избр. соч., т. 1, М., 1965; Агрохимия, под ред. В. М. Клечковского и А. В. Петербургского, М.; 1967; Применение органических удобрении, М. 1971. И. П. Мамченков. Органический мир Органи'ческий мир,совокупность живых существ - , населяющих Земли. О. м. слагается из микроорганизмов, растений, животных и человека (см. , , раздел История развития органического мира). Отдельные компоненты О. м. в процессе жизнедеятельности тесно взаимодействуют между собой и с телами неорганической (косной) природы. См. также , . Органический статут Королевства Польского 1832 Органи'ческий стату'т Короле'вства По'льского 1832,конституционный статут Королевства Польского, введённый после подавления вместо конституции 1815. Издан 14(26) февраля 1832 в Петербурге Николаем I. Упраздняя существовавшие ранее элементы польской государственности (сейм, отдельную польскую армию и др.), О. с. К. П. сохранял ряд автономных учреждений (наместничество, Государственный совет и Совет управления при наместнике и др.), которые по мере распространения на территории Королевства Польского административных норм Российской империи постепенно ликвидировались. В 1861, в условиях начавшегося революционного подъёма, царское правительство было вынуждено восстановить некоторые учреждения, предусмотренные О. с. К. П. (например, Государственный совет, ликвидированный в 1841). Органическое вещество Органи'ческое вещество',обязательный малый компонент почв, поверхностных и подземных вод, большинства горных пород, особенно осадочных, а также атмосферы (см. рис. ). Первоисточником О. в. являются в основном растения и в значительно меньшей степени животные. По первоначальному составу различают три основных типа О. в. Гумусовое О. в. образовалось преимущественно из высших растений и различных обитателей почвы (главным образом микроорганизмов); слагает большую или меньшую часть почв, основную массу и , отношение Н/С обычно меньше 0,9. Сапропелевое О. в. - продукт изменения низших растений (главным образом планктонных водорослей), свойственно , и рассеянному О. в.; Н/С обычно больше 1,2. Липтобиолитовое О. в. возникает за счёт концентрации химически наиболее стойких компонентов растительные вещества. Известны и др. типы О. в., образовавшиеся благодаря существованию в прошлом некоторых групп животных организмов (например, остатки граптолитов и ряда ракообразных, состоящие из хитина). Как рассеянное в осадочных породах, так и концентрированное О. в. ( ) проходит (при ) ряд стадий - от буроугольной (в зоне с температурой до 60-70 °С) до антрацитовой (в зоне с температурой более 250 °С). В конечном счёте, в зоне (метаморфизма) О. в. переходит в графит. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 |
|||||||||||||||||